







Pulmonary hypertension is classified into five groups by the World Health Organization (WHO). Group 1, pulmonary arterial hypertension (PAH), is a progressive disease characterized by an elevation in pulmonary arterial pressure and pulmonary vascular resistance that may progress to right heart dysfunction and failure.1 PAH is defined as a mean pulmonary artery pressure (mPAP) ≥25 mmHg at rest, with a pulmonary capillary wedge pressure ≤15 mmHg and a pulmonary vascular resistance (PVR) >3 Wood units measured by right-sided cardiac catheterization.2–4
PAH is associated with certain medical conditions or as an idiopathic disease (idiopathic PAH [IPAH]). The prevalence of IPAH is estimated to be 5.9 cases per million adults in North America and Europe, with a female predominance (male-to-female ratio, 1:1.7).1–4 Recent registry data reveal that PAH is now being diagnosed more commonly in older patients, with a mean age at diagnosis ranging from 50 to 65 years old.1,2,4 Medical conditions associated with PAH include certain congenital heart diseases; rheumatologic diseases including but not limited to scleroderma, systemic lupus erythematosus, and rheumatoid arthritis; human immunodeficiency virus (HIV) infection, and portal hypertension.3 The contributing cause of PAH can be important for both outcomes and management. For example, patients with scleroderma who develop PAH, estimated between 7 % and 12 % of patients, have markedly worse outcomes in comparison to other PAH subgroups.3
Multiple drugs and toxins have been associated with PAH including anorexigens such as fenfluramine and dexfenfluramine, toxic rapeseed oil, and even illicit drugs such as cocaine and amphetamines.1,3,4 Selective serotonin reuptake inhibitors (SSRIs) increase the risk of persistent pulmonary hypertension in newborns of pregnant women receiving SSRIs.4 Heritable PAH (HPAH) includes both IPAH with germline mutations and familial cases without an identified mutations, however, about 75 % of patients with HPAH have a bone morphogenetic protein receptor 2 (BMPR2) mutation.2
Epidemiology and Pathophysiology
The prevalence of PAH is estimated to be 15–26 patients per million individuals, with only 15,000–20,000 of these patients currently receiving treatment.5 The US-based REVEAL registry (Registry to Evaluate Early and Long-Term Pulmonary Arterial Hypertension Disease Management) includes over 3,500 patients and found that 46 % of PAH was idiopathic while 25 % was associated with connective tissue diseases and 10 % was associated with congenital heart diseases.6 Early diagnosis of PAH is improving due to increased awareness and knowledge of the disease state. IPAH is associated with a poor prognosis (median survival 2.8 years) after diagnosis without targeted therapy.7 Prior to the availability of disease-specific therapy for IPAH, survival rates for 1, 3, and 5 years were 68 %, 48 %, and 34 %, respectively.8 A recent epidemiologic study demonstrated survival rates at 1 and 3 years were 85 % and 68 %, respectively, in patients with PAH, and 91 % and 74 %, respectively, in patients with IPAH.9
PAH is caused by progressive vasoconstriction of the small pulmonary arteries. All subgroups of PAH have similar clinical and pathologic physiology, including endothelial cell dysfunction, thrombotic lesions, platelet activation, gain of constricting factors, loss of relaxing factors, intimal proliferation, medial hypertrophy, fibrosis, and inflammation.10 Progressive vascular remodeling occurs, leading to increased pulmonary arterial pressures and pulmonary vascular resistance. The right ventricle is normally exposed to much lower pressures than the left ventricle. In the setting of persistent elevations in pulmonary pressures, right ventricular hypertrophy and eventually right heart failure develop.11
Molecular, cellular, and genetic mechanisms that contribute to PAH are affected by several compounds, including prostacyclin (PGI2), endothelin-1 (ET-1), and nitric oxide (NO). PAH is characterized by decreased NO synthase expression and levels of circulation PGI2, a vasodilatory and antiproliferative substance that is produced by the endothelial cells, and increased levels of potent vasoconstrictors, including thromboxane and ET-1. 10,12,13 Plasma levels of ET-1 are correlated with severity of PAH and prognosis.14
Clinical Presentation and Assessment of PAH
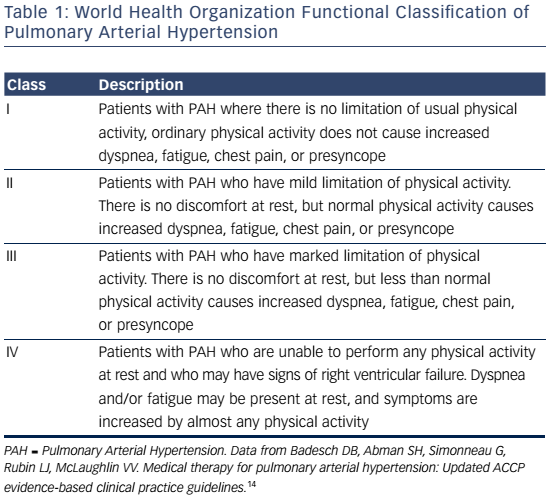
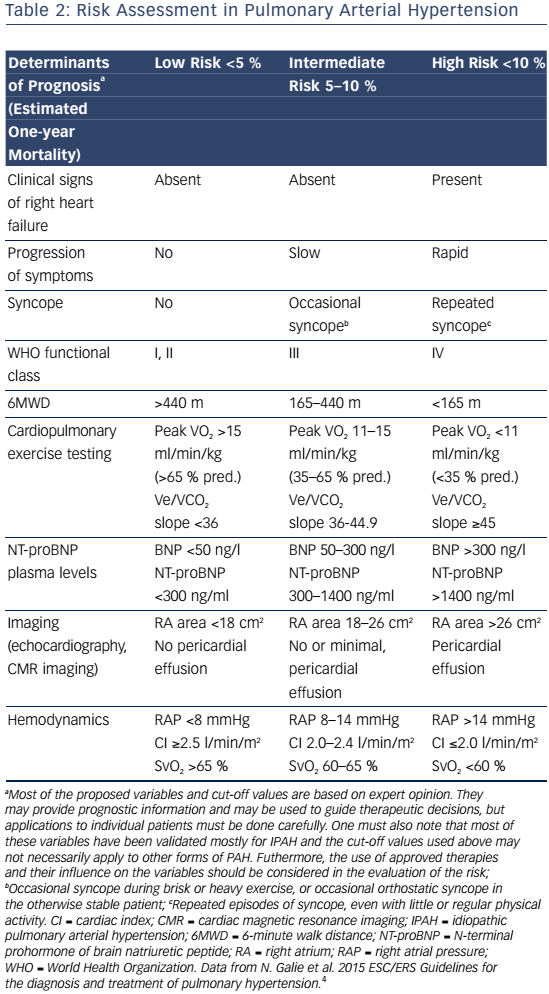
Signs and symptoms of PAH vary depending on disease severity and comorbidities. As right ventricular dysfunction progresses, patients may experience exertional dyspnea, fatigue, weakness, syncope, lower extremity edema, and abdominal bloating and distension.4 Assessment of functional class is used to determine the impact of signs and symptoms on the patient function and quality of life (QoL).Table 1 shows patients with an increased risk of mortality are more likely to have a higher WHO functional class, older age, male gender, higher brain natriuretic peptide (BNP), higher right atrial pressure, and lower cardiac output. In contrast, patients with a decreased risk of mortality are more likely to have a lower WHO functional class, higher 6-minute walk distance (6MWD), lower BNP, and higher cardiac output.15
Doppler echocardiography serves as a noninvasive screening test to detect increased pulmonary pressures, and, more importantly, the morphology of the right-sided heart chambers. It can also be used to identify any left-sided heart disease or congenital heart disease.16 However, right heart catheterization (RHC) is required to definitively diagnose PAH. Pulmonary vasoreactivity is assessed during the RHC in specific subgroups of PAH, including IPAH, HPAH, or drug-induced PAH.17 A positive vasoreactivity response is a reduction of mPAP by at least 10 mmHg to a value of 40 mmHg or less with a stable cardiac index.18 Patients with a positive acute response (approximately 13 % of patients on initial testing) are most likely to have a beneficial hemodynamic and clinical response. Unfortunately, about half of these patients have negative response when tested 1 year later.19
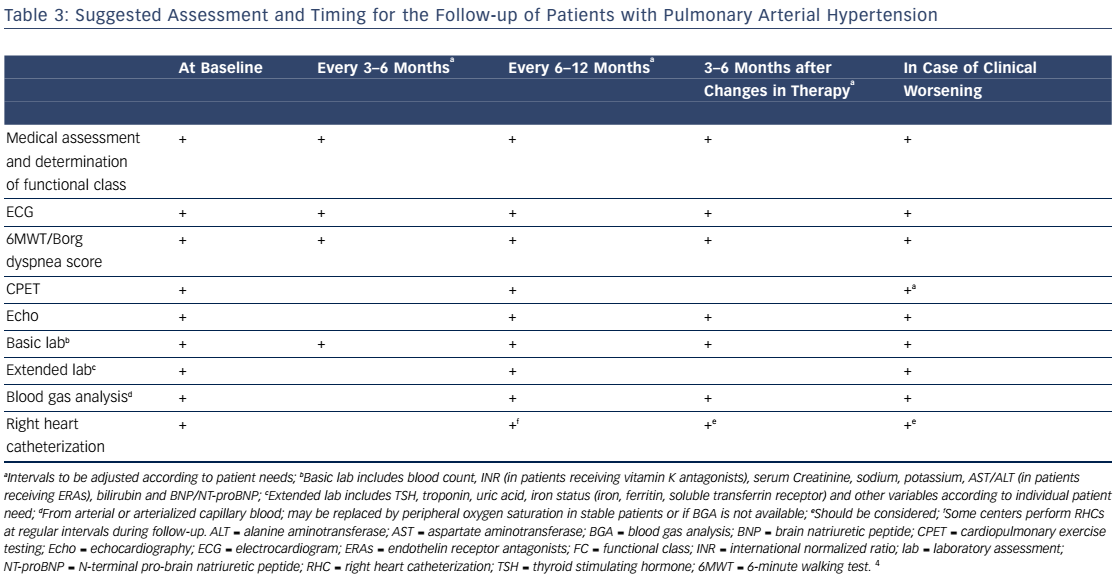
PAH commonly occurs due to connective tissue disease so serologic markers should be obtained to confirm or exclude these diagnoses.3,20 In addition, liver function tests, HIV status, pulmonary function tests, ventilation-perfusion lung scans and/or pulmonary angiography, and arterial blood oxygenation should all be assessed when establishing a PAH diagnosis and cause.3 Functional class and exercise capacity, determined by the 6MWD, should be assessed at baseline and at regularly scheduled clinic visits. Assessment of these and other variables can help categorize patient risk (shown in Table 2).3,20Table 3 provides additional information on initial assessment and timing and when each assessment is indicated.
Approach to Treatment
The goal of treatment of PAH is to improve functional class, exercise capacity, and QoL while delaying disease progression. Patients with PAH should be referred to an expert, specialized PAH center for early assessment of hemodynamics on right heart catheterization and optimal management. Treatment of PAH may be categorized into nonpharmacologic, conventional therapy, disease-specific therapy, and surgical interventions. Surgical therapy includes atrial septostomy, pulmonary thromboendarterectomy for CTEPH, and lung or heart–lung transplantation for severe disease.
Nonpharmacologic Therapy
Nonpharmacologic therapy may address comorbid conditions that often accompany PAH. Counseling of avoidance of pregnancy is key due to high morbidity and mortality in females with PAH during and after pregnancy.7 All patients should receive immunization against influenza and pneumococcal disease.3 Certain PAH patients may require supplemental oxygen to keep oxygen saturations to a goal of greater than 90 % and to decrease hypoxic vasoconstriction. It is important to remember when in high altitude or air travel that there is a reduction in ambient air concentration of oxygen.21 Patients should maintain a low-sodium diet to avoid fluid retention in right heart failure.22 Cardiopulmonary rehabilitation improves functional status, exercise capacity, and QoL in patients with PAH.1
Pharmacologic Therapy
Conventional Pharmacologic Treatment
Conventional therapy includes oral anticoagulants, diuretics, and digoxin.20 Anticoagulation with warfarin (goal international normalized ratio [INR] 1.5–2.5) may be considered in patients with PAH, particularly if they have IPAH, HPAH, or PAH due to anorexigens, to prevent in situ thrombosis of pulmonary arteries and decrease the risk of venous thromboembolism.4 Small retrospective and prospective studies support a survival benefit with anticoagulation.23–26 Anticoagulation is not recommended for patients with PAH associated with portal hypertension or HIV due to increased risk of bleeding.4 Loop diuretics are indicated in patients with decompensated right heart failure and hypervolemia as indicated by increased central venous pressure, abdominal organ congestion, peripheral edema, and ascites.3 Digoxin may be useful in certain patients with PAH as adjunctive therapy to diuretics in management of right heart failure and in patients with atrial arrhythmias.1 The typical target concentration is between 0.5 and 0.8 ng/mL.
Calcium Channel Blockers
Calcium channel blockers (CCBs) are infrequently used in the management of PAH and can only be used in patients with a positive response to acute vasodilator testing. As noted above, there are very few patients who demonstrate this response (approximately 13 % of patients with IPAH and even less with other etiologies of PAH). The number responding to long-term therapy is even lower (7 %). 19 Vigilant monitoring to ensure adequate and continued response is required when CCBs are used. CCBs should not be used in the absence of demonstrated acute vasoreactivity.1
Disease-Specific Pharmacologic Therapy
Therapeutic targets in the treatment of PAH focus on supplementing endogenous vasodilators, inhibiting endogenous vasoconstrictors, and reducing endothelial platelet interaction and limiting thrombosis. Five classes of disease-specific agents for PAH are now available: prostacyclin analogs, endothelin receptor antagonists, phosphodiesterase inhibitors, a soluble guanylate cyclase stimulator, and a prostacyclin IP receptor agonist.
Synthetic Prostacyclin and Prostacyclin Analogs
PGI2 is produced by endothelial cells and is a potent vasodilator of all vascular beds. In addition, it also inhibits platelet aggregation and has both cytoprotective and antiproliferative properties. However, in patients with PAH, PGI2 synthase expression is reduced in pulmonary arteries, leading to pulmonary vasoconstriction and increased platelet aggregation.27 Several prostacyclin analogs are now available for treatment of PAH, including epoprostenol (available as IV Flolan® and Veletri®), treprostinil (IV/SC: Remodulin®, inhaled: Tyvaso®, oral: Orenitram®), and iloprost (Ventavis®).
Epoprostenol is a synthetic analog of PGI2 that was introduced in 1995 as the first disease-specific therapy for patients with PAH, indicated for patients with WHO functional class III or IV. A 12-week, openlabel, randomized trial comparing continuous infusion epoprostenol to conventional therapy in functional class III and IV IPAH patients demonstrated improved exercise capacity, QoL, hemodynamics, and functional class in patients treated with epoprostenol. Despite the short trial duration of 12 weeks, investigators also found an improvement in survival in patients treated with epoprostenol versus conventional therapy (0 versus 8 patients, respectively; P=0.003); this finding remained significant after adjustment for a numerical difference in 6MWD at baseline (P<0.002).28
In addition, observational studies also support improved survival in patients with IPAH on epoprostenol compared with either historical control or predicted survival based on the National Institutes of Health Registry equation. 29–31 It has a very short half-life of 3–5 minutes, requiring administration by continuous IV infusion. Epoprostenol must be initiated in the hospital setting at a low dose (2–4 ng/kg/min) and titrated up based on tolerance of side effects such as flushing, headache, diarrhea, jaw pain, abdominal cramping, and hypotension. The target dose for the first 2–4 weeks is around 10–15 ng/kg/min. Patients are then titrated up based on how well they respond and side effects.21,29 The two available products, Flolan® and Veletri®, are unique in terms of stability and reconstitution with Flolan® being unstable at room temperature and Veletri® being the thermostable formulation. Patients must always have a backup supply of the drug and infusion pump as interruption of epoprostenol may lead to life-threatening pulmonary vasoconstriction.32 Infection, catheter obstruction, and sepsis are potential complications due to the route of administration.33
Treprostinil (Remodulin®) is a stable analog of PGI2 given for subcutaneous (SC) or IV infusion for WHO functional class III and IV patients.29 Notable advantages of treprostinil over epoprostenol include ease of use given stability at room temperature and increased safety due to a longer half-life (about four hours), lowering the risk of rebound pulmonary vasoconstriction that may happen with drug interruption.12 Treprostinil improves 6MWD and hemodynamics with outcomes that are similar to epoprostenol.34,35 A 12-week double-blind, placebo-controlled trial of 470 patients with WHO functional class II, III or IV PAH demonstrated that patients receiving SC treprostinil versus placebo had significant improvement in exercise capacity, dyspnea, and hemodynamic parameters.
Exercise capacity improved most in patients with a lower baseline 6MWD and in patients who could tolerate doses greater than 13.8 ng/kg/min.34 The initial dose for treprostinil is 1.25 ng/kg/min SC. Infusion site pain is common with the SC route, occurring in up to 85 % of patients and leading to discontinuation in 8 % of patients.21 Intravenous treprostinil may be considered in patients unable to tolerate SC.27 Transitions between prostacyclin agents or routes should be performed in an inpatient setting. Bacteremia, primarily due to gram-negative pathogens, have been reported more commonly with IV treprostinil than with IV epoprostenol.36
Two inhaled formulations were developed in an effort to prevent complications associated with parenteral prostacyclin administration. Iloprost (Ventavis®) is indicated for WHO functional class III and IV patients based on a 12-week randomized, placebo-controlled trial of 203 patients. Significantly more patients receiving inhaled iloprost versus placebo met the combined primary endpoint of improvement by one functional class and improvement in 6MWD by 10 % (16.8 % versus 4.9 %, respectively; P=0.007).37 In addition, the placebo group had more clinical deterioration over the study duration (13.7 % with placebo versus 4.0 % with iloprost; P=0.024).37 It is given by inhalation using a dosing system provided by the manufacturer (ADD system) with the initial dose being 2.5 mcg 6–9 times per day up to every 2 hours during waking hours. The dose should be titrated and maintained at 5 mcg/dose if tolerated. While clinical trials with iloprost have demonstrated an improvement in exercise capacity and functional class, it can be cumbersome to use as each inhalation dose can take 4–10 minutes to administer and multiple inhalations are required for a full dose.37 A backup supply is also necessary with iloprost due to a short half-life, similar to epoprostenol.18 Adverse effects are similar to other PGI2 analogs, including headache, flushing, and jaw pain. Inhaled treprostinil may also cause throat irritation and cough.
Inhaled treprostinil (Tyvaso®) improves exercise capacity and functional class and may be used for WHO functional class III and IV patients. In a randomized, double-blind, 12-week trial, patients receiving inhaled treprostinil in addition to bosentan or sildenafil experienced a 20-meter improvement in 6MWD compared to placebo (P<0.0006).38 A 2-year extension of the trial found that inhaled treprostinil provided sustained benefit and remained safe and efficacious.39 The approved initial dosing of inhaled treprostinil is three breaths (6 mcg per breath) four times daily during waking hours, and this dose should be titrated based on patient tolerance at 1- to 2-week intervals to maximum dose of nine breaths (total of 54 mcg) four times daily. Inhaled treprostinil requires less time to administer than iloprost but may be more complicated for patients to prepare.18 Common adverse effects include throat irritation and cough (unique to the inhaled products), headache, nausea, dizziness, and flushing. Inhaled treprostinil may also cause systemic hypotension, requiring careful monitoring if patients are concurrently on diuretics, antihypertensives, or other vasodilators.
The first oral prostacyclin analog, sustained-release trepostinil (Orenitram®), was approved by in the US in 2013 for patients with functional class II and III PAH. Oral treprostinil monotherapy improved 6MWD, but not functional class or time to clinical worsening, in a 12-week study of 349 patients with PAH.40 Oral treprostinil has also been studied in combination with endothelin receptor antagonists and/ or phosphodiesterase-5 inhibitors, without improvement in 6MWD.41,42 However, patients receiving higher doses of treprostinil did show more improvement in exercise capacity as measured by 6MWD. Adverse events include headache, nausea, diarrhea, and jaw pain. Oral treprostinil, like all prostacyclin analogs, inhibits platelet aggregation and may increase bleeding risk, particularly in patients receiving oral anticoagulants.
Endothelin Receptor Antagonists
Three Endothelin Receptor Antagonists (ERAs) are currently available for management of patients with WHO functional class II, III, or IV PAH: bosentan (Tracleer®), ambrisentan (Letairis®), and macitentan (Opsumit®). Bosentan is an orally active dual ETA and ETB receptor antagonist. In a 16-week randomized, placebo-controlled, double-blind trial (BREATHE-1) enrolling 213 patients, those receiving bosentan 125 mg twice daily (BID) or 250 mg BID versus placebo had improved exercise capacity, functional class, hemodynamics, echocardiographic, and Doppler variables, and time to clinical worsening.43 The initial dose is 62.5 mg twice daily for 4 weeks followed by 125 twice daily.43 Increased hepatic transaminases may occur due to competition by bosentan and its metabolites with the biliary excretion of bile salts, resulting in retention of bile salts that are cytotoxic to hepatocytes. In clinical trials, bosentan 250 mg BID was associated with a dose-dependent increase in hepatic transaminases that occurred significantly more than with the 125 mg BID dose. For this reason, bosentan 125 mg oral BID is the US Food and Drug Administration (FDA)-approved dose and the drug is only available through a distribution program, the Tracleer Access Program.18 Vigilant monitoring of liver function tests (LFTs) is required at baseline and every month, and dose reduction or interruption may be necessary if LFTs increase. Complete blood cell count should be monitored every 3 months due monitor for anemia while on bosentan. For all three of the ERAs, monthly pregnancy testing is required in women (pregnancy category X).
Ambrisentan is a once-daily selective ETA receptor antagonist that improves exercise capacity and hemodynamics and delays clinical worsening in PAH.44 Two large, concurrent randomized, doubleblind, placebo-controlled trials: Ambrisentan in Pulmonary Arterial Hypertension, Randomized, Double-Blind, Placebo-Controlled, Multicenter, Efficacy Study 1 and 2 (ARIES-1 and ARIES-2) over 12 weeks demonstrated a significant improvement in exercise capacity with 2.5, 5, and 10 mg daily doses.45 Most patients had WHO functional class II or III PAH. A 2-year extension trial (ARIES-E) supports long-term efficacy of ambrisentan with results showing continued improvement in 6MWD and functional class.46 Similar to bosentan, ambrisentan is only available through a distribution program, Letairis Education and Access Program (LEAP).18 Unlike bosentan, liver toxicity occurs very rarely with ambrisentan (0.8 % in 12-week trials and 2.8 % for up to 1 year).45,46 Common side effects include peripheral edema, nasal congestion, flushing, anemia, and palpitations. Treatment should be initiated with 5 mg once daily and increased to 10 mg once daily if required.
Macitentan is a once-daily dual ERA recently approved in the US following the Study with an Endothelin Receptor Antagonist in Pulmonary Arterial Hypertension to Improve Clinical Outcome (SERAPHIN) trial.47 Over approximately 3 months, both doses (3 and 10 mg) demonstrated statistically significant decreases in the composite end point of events related to PAH or death compared to placebo. Worsening of PAH was the most common event (defined as a decrease in 6MWD, worsening symptoms, and need for additional treatment). Patients were continued on concomitant therapy, if at stable doses for 3 months, with oral or inhaled prostanoids, calcium channel blockers, or oral phosphodiesterase inhibitors. Increases in LFTs were similar to placebo. The FDA approved dose of macitentan is 10 mg daily. The most common side effects include nasopharyngitis, headache, and anemia. Female patients must go through a Risk Evaluation and Mitigation Strategy (REMS) program to receive the drug.
Phosphodiesterase-5 Inhibitors
Two phosphodiesterase-5 inhibitors available for the treatment of WHO functional class II and III PAH, sildenafil (Revatio®) and tadalafil (Adcirca®), work by increasing the intracellular concentration of cyclic guanosine monophosphate, leading to vasorelaxation and antiproliferative effects on vascular smooth muscle cells. Sildenafil is a potent and highly specific phosphodiesterase-5 inhibitor shown to reduce mPAP and improve 6MWD, hemodynamic parameters and functional class.48 In a double blind, placebo-controlled trial, Sildenafil Use in Pulmonary Arterial Hypertension (SUPER), of 278 primarily WHO functional class II and III patients, those receiving sildenafil showed a significant improvement in 6MWD and hemodynamics at 12 weeks versus placebo. A continued improvement in 6MWD was seen in a 1-year extension study.48
The FDA-approved dose is 20 mg by mouth three times per day, however much higher doses, up to 80 mg three times daily, have been used. Adverse effects include headaches, flushing, epistaxis, dyspepsia, hypotension, and diarrhea. Changes in vision have been reported, including blue-tinted vision and sudden loss of vision, requiring drug discontinuation. Tadalafil was approved by the FDA in 2009 following a 16-week double-blind, placebo-controlled study of 405 patients;
Pulmonary Arterial Hypertension and Response to Tadalafil (PHIRST). Patients primarily had WHO functional class II and III PAH and 53 % were on background therapy of bosentan. Use of tadalafil 40 mg daily was shown to significantly improve exercise capacity, QoL measures, and time to clinical worsening.49 Tadalafil and sildenafil levels are decreased in patients on bosentan therapy due CYP450 3A4 induction, requiring higher doses of both phosphodiesterase-5 inhibitors in patients on concurrent bosentan therapy.18,48 The most commonly reported adverse events were headache, myalgia, and flushing.49 Concurrent use with nitrate therapy is contraindicated and must be avoided with both sildenafil and tadalafil due to additive hypotension.
Soluble Guanylate Cyclase Stimulator
Riociguat (Adempas®) is a soluble guanylate cyclase stimulator that works synergistically with nitric oxide to increase pulmonary vasolidation. Riociguat was approved in 2013 for patients with WHO functional class II, III, and IV PAH following the Pulmonary Arterial Hypertension Soluble Guanylate Cyclase-Stimulator Trial 1 (PATENT-1) trial, which demonstrated that riociguat 2.5 mg by mouth three times daily significantly improved 6MWD, hemodynamic parameters, and WHO functional class compared to placebo.50 Baseline therapy of endothelinreceptor antagonists or nonintravenous prostacyclin analogs were continued. Importantly, use of riociguat with phosphodiesterase-5 inhibitors and nitrates is contraindicated due to the additive risk of hypotension. The initial dose is 1 mg by mouth three times daily, titrated by 0.5 mg three times daily every 2 weeks to a maximum dose of 2.5 mg by mouth three times daily Like the ERAs, riociguat is teratogenic and female patients must go through a REMS program to receive the drug.
Prostacyclin IP Receptor Agonist
Selexipag (Uptravi®), a novel prostacyclin IP receptor agonist, has been shown to decrease disease progression, hospitalizations for PAH, and complications from PAH, including death.51 In a phase 3, double-blind, placebo-controlled trial: Selexipag for the Treatment of Pulmonary Arterial Hypertension (GRIPHON) of 1156 patients with WHO functional class II and III, selexipag was associated with a decrease in death from any cause or complications related to PAH versus placebo (27.0 % versus 41.6 %, respectively; HR 0.60, 99 % CI [0.46–0.78]; P<0.001). Patients were followed for up to 3 years on therapy. These outcomes were similar in patients on no background therapy and when added to background ERAs, phosphodiesterase-5 inhibitors, or both.51 The initial starting dose is 200 mcg PO twice daily; this dose can be increased by 200 mcg twice daily increments to a maximum dose of 1600 mcg twice daily. Common side effects are similar to those caused by prostacyclin analogs, including flushing, headache, diarrhea, nausea, jaw pain, and myalgias. Selexipag may also cause anemia so a complete blood cell count should be monitored periodically. Selexipag has also been associated with an increased incidence of hyperthyroidism.
Combination Therapy
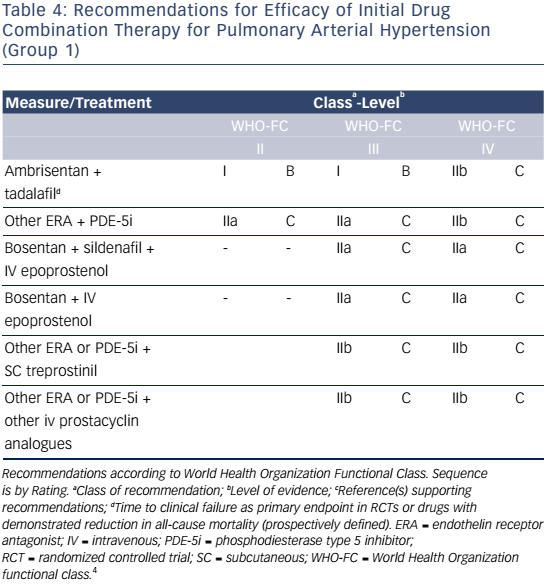
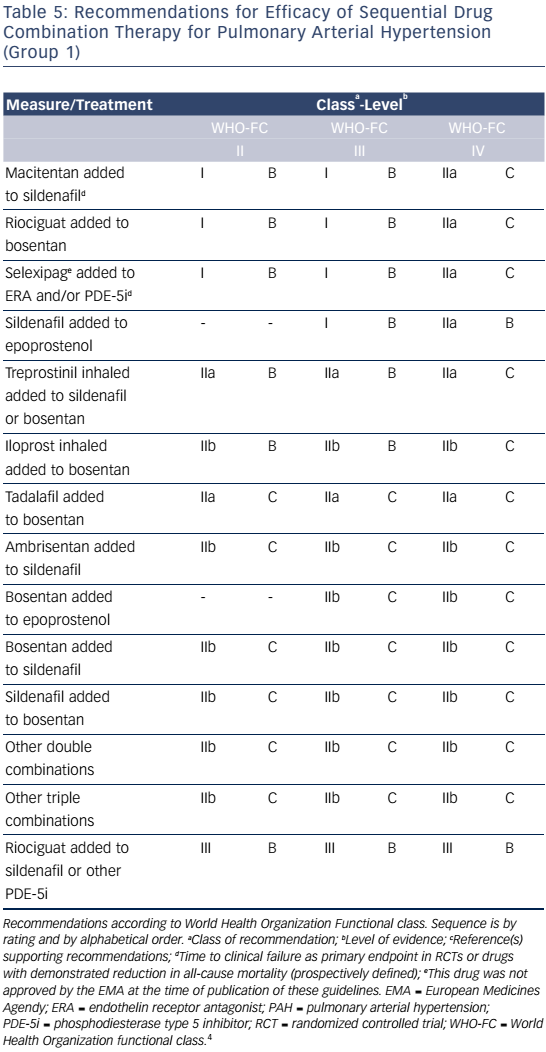
Combination therapy can target multiple pathophysiologic mechanisms in PAH, resulting in improvement in hemodynamics, symptoms, functional class, and exercise capacity.52–55 Combination therapy may be started sequentially as PAH progresses or as initial therapy. Recent evidence-based guidelines provide comprehensive tables detailing drug combinations that have been studied for both initial combination therapy (see Table 4) and sequential combination therapy (see Table 5). The Initial Use of Ambrisentan plus Tadalafil in Pulmonary Arterial Hypertension (AMBITION) trial comparing ambrisentan and tadalafil together versus either alone in WHO functional class II or III demonstrated that initial combination therapy was associated with a significant reduction in time to clinical failure and PAH hospitalizations. Adverse effects such as peripheral edema, headache, nasal congestion, and anemia were more common in the combination group than either monotherapy group, although discontinuation rates were similar.56
Evaluation of Therapeutic Outcomes
Response to therapy should be followed closely. Monitoring involves objectively assessing functional status and exercise capacity with the 6MWD and hemodynamics and right ventricular function with echocardiography and right heart catheterization. It is also very important to monitor subjective measures like WHO functional class and quality of life outcomes.Table 3 provides suggested recommendations for specific baseline and follow-up assessments and when each is indicated.
Conclusion
Significant advances have been made in determining the pathogenesis of PAH as well as in the evaluation and treatment of these patients over the past three decades. With approved targeted therapies such as ERAs, phosphodiesterase-5 inhibitors, and PGI2 analogs, clinical improvement is possible in most patients, leading to a better QoL and delay of disease progression. Recent guidelines provide clinicians recommendations for the initiation of combination therapy as well as recommendations for monitoring and assessment of patient response.